Medaka (Oryzias latipes) has become an important vertebrate model widely used in genetics, developmental biology, environmental sciences, and many other fields. A high-quality genome sequence and a variety of genetic tools are available for this model organism. However, existing genome annotation is still rudimentary, as it was mainly based on computational prediction and short-read RNA-seq data. The low-quality genome annotation remains a major obstacle for omics studies in medaka.
Recently, a team lead by Dr. TU Qiang at the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, collaborated with Dr. Kiyoshi Naruse at Japan National Institute for Basic Biology, reported a multi-omics landscape of medaka embryogenesis profiled by long-read RNA-seq, short-read RNA-seq, and ATAC-seq. This work constructed a much-improved gene model set including about 17,000 novel isoforms and identified 1600 transcription factors, 1100 long non-coding RNAs, and 150,000 potential cis-regulatory elements as well.
Time-series datasets provided another dimension of information. They obtained the temporal expression profiles of all these genes during embryogenesis, and identified those that employ different isoforms across developmental stages. With the expression dynamics of genes and accessibility dynamics of cis-regulatory elements, they investigated regulatory logic between accessible elements and genes during embryogenesis. For cis-regulatory logic between genes and their nearby DNA elements, they found at least four common types: synchronization, repression, enhancer switching, and early opening. For trans-regulatory logic between genes and TFs, they found multiple pioneer TFs in early embryogenesis.
Ultimately, the research team built a user-friend medaka omics data portal (http://tulab.genetics.ac.cn/medaka_omics) to present the data alongside other published genomics datasets. Researchers can easily search for various genomics tracks for embryonic stages and adult tissues samples, accrued via different omics technologies. Users can also search for genes with various annotation information and make quantification plots of gene expression.
The work provides the first comprehensive omics datasets of medaka embryogenesis. The data portal will serve as a daily reference tool for the entire medaka community. Besides, the authors term three omics assays as the minimum ENCODE toolbox and propose the use of it as the initial and essential profiling genomic assays. The use of this toolbox will be valuable for a large number of model organisms that have limited omics data available.
The paper entitled “
Dynamic Transcriptional and Chromatin Accessibility Landscape of Medaka Embryogenesis” was published online in
Genome Research on June 26, 2020 (
DOI:10.1101/gr.258871.119). This work was supported by the National Natural Science Foundation of China and the Strategic Priority Research Program of the Chinese Academy of Sciences.
Figure: Multi-omics landscape of medaka embryogenesis (Image by IGDB). (a) The improved annotation of the medaka genome. (b) Cis-regulatory logic between genes and accessible elements: synchronization, repression, enhancer switching, and early opening. (c) Trans-regulatory logic between genes and TFs: pioneer TFs identified at stage 11. (d) Medaka omics data portal.
Contact:
Dr. TU Qiang
Institute of Genetics and Developmental Biology, Chinese Academy of Sciences
 CAS
CAS
 中文
中文



.png)
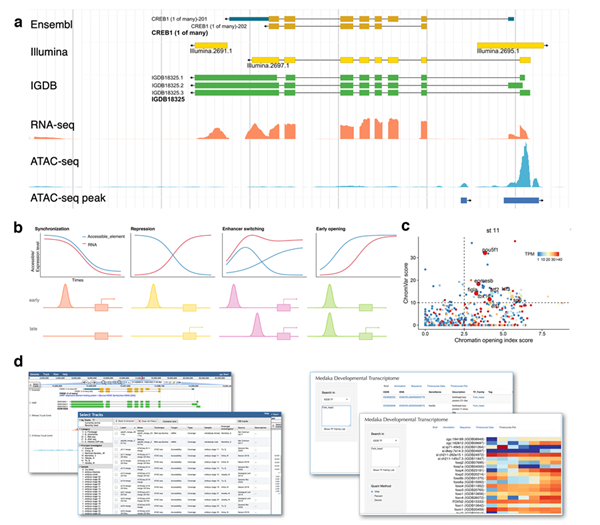 Figure: Multi-omics landscape of medaka embryogenesis (Image by IGDB). (a) The improved annotation of the medaka genome. (b) Cis-regulatory logic between genes and accessible elements: synchronization, repression, enhancer switching, and early opening. (c) Trans-regulatory logic between genes and TFs: pioneer TFs identified at stage 11. (d) Medaka omics data portal.Contact:Dr. TU QiangInstitute of Genetics and Developmental Biology, Chinese Academy of SciencesEmail: qtu@genetics.ac.cn
Figure: Multi-omics landscape of medaka embryogenesis (Image by IGDB). (a) The improved annotation of the medaka genome. (b) Cis-regulatory logic between genes and accessible elements: synchronization, repression, enhancer switching, and early opening. (c) Trans-regulatory logic between genes and TFs: pioneer TFs identified at stage 11. (d) Medaka omics data portal.Contact:Dr. TU QiangInstitute of Genetics and Developmental Biology, Chinese Academy of SciencesEmail: qtu@genetics.ac.cn