Scientists at the Institute of Genetics and Developmental Biology of the Chinese Academy of Sciences developed a simplified and highly efficient 3’RNA-seq method, called Simplified Poly(A) Anchored Sequencing (SiPAS), to boost population transcriptomics in plants. The study was recently published in
Plant Biotechnology Journal on September 12, 2021 (
https://onlinelibrary.wiley.com/doi/10.1111/pbi.13706).
RNA sequencing (RNA-seq) is a keystone technology for modern biological research, shifting many genomic studies from a solely genomic level to a multi-omic level and thus effectively improving our understanding of genome biology. Over the last few years, vast amounts of genomic data have been generated in many plant species. The gigantic size of genomic data is creating a vacuum where a large quantity of transcriptomic data needs to be filled to help decode the function of the genome. Highly efficient RNA-seq technologies are increasingly demanded in biological research.
The emergence of 3’RNA-seq is a giant leap of RNA-seq technologies. Active methodological development has been made to the 3’RNA-seq technology. However, these 3’RNA-seq methods usually use custom sequencing format and have not been suf?ciently optimized for the standard paired-end 150/250 bp (PE150 or PE250) sequencing. This hinders their application in large-scale population transcriptomic study.
In this study, WANG Jing, XU Jun and YANG Xiaohan et al. in LU Fei’s research group developed an efficient gene expression profiling approach, Simplified Poly(A) Anchored Sequencing (SiPAS), by combining the advantages of reported 3’RNA-seq methods and optimizing the use of standard PE150 sequencing format. Through testing SiPAS for its performance in bread wheat (Triticum aestivum. ssp. aestivum, 2n = 6x = 42, genome size = 16 Gb), they presented evidence showing that SiPAS achieves a high level of sensitivity, accuracy, and reproducibility.
The improved 3’ RNA-seq method SiPAS provides multiple strengths to advance population transcriptomic studies in plants. First, SiPAS is effortless and cost-effective with the simplified workflow. Meanwhile, SiPAS is optimized and well suited for the standard sequencing format of Illumina (PE150). Benefiting from the simplified and standardized library construction process, the cost of SiPAS is substantially reduced to $2/sample. Second, SiPAS is highly effective in quantifying gene expression. By switching P5 and P7 adapters, the read end used for alignment achieves higher base quality, resulting in increased read mapping sensitivity and a high level of accuracy and reproducibility of gene expression quantification. Notably, for 107,891 genes in the wheat genome, only five million reads achieved Pearson’s r of 0.96 between the gene expression level of two technical replicates. This suggests that SiPAS may not require technical replicates for transcriptomic analysis when the sample size is large. Third, SiPAS is robust to RNA degradation. This is because the 3’RNA is generally more stable than the rest of RNA sequences. The high tolerance to RNA degradation lessens the variability during sample preparation and guarantees a fair comparison of gene expression between samples.
Overall, with the strengths of being cost-effective and laborsaving, and equivalent performance with TruSeq, SiPAS promises the ease of use of this method in large-scale population transcriptomic studies. We anticipate that SiPAS will be used in many species and contribute to an in-depth understanding of plant genomes.
Figure 1 Comparison of SiPAS with TruSeq. (Image by IGDB)
Figure 2 The performance of SiPAS on degrading RNA library. (Image by IGDB)
Contact:
Dr. LU Fei
Institute of Genetics and Developmental Biology, Chinese Academy of Sciences
 CAS
CAS
 中文
中文



.png)
 Figure 1 Comparison of SiPAS with TruSeq. (Image by IGDB)
Figure 1 Comparison of SiPAS with TruSeq. (Image by IGDB)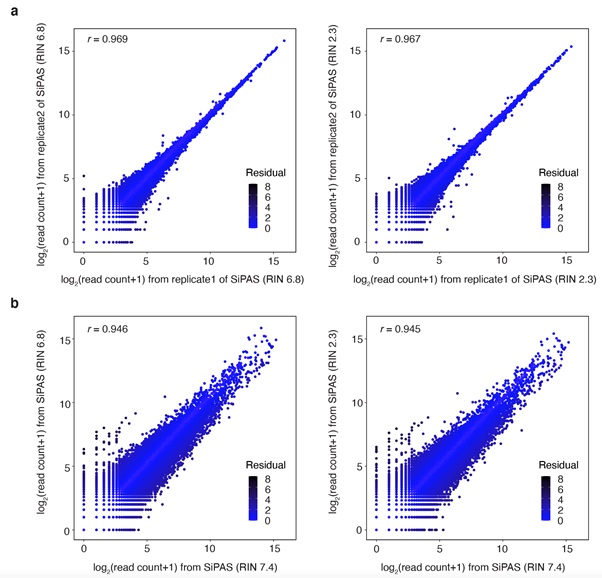 Figure 2 The performance of SiPAS on degrading RNA library. (Image by IGDB)Contact:Dr. LU FeiInstitute of Genetics and Developmental Biology, Chinese Academy of SciencesEmail: flu@genetics.ac.cn
Figure 2 The performance of SiPAS on degrading RNA library. (Image by IGDB)Contact:Dr. LU FeiInstitute of Genetics and Developmental Biology, Chinese Academy of SciencesEmail: flu@genetics.ac.cn