Pyridoxine-dependent epilepsy (PDE) is a rare autosomal recessive genetic disorder characterized by recurrent seizures in neonates. PDE is caused by mutations in the Aldh7a1 gene, leading to a deficiency in a key enzyme in lysine metabolism and the accumulation of toxic metabolites AASA/P6C. Clinical observations show that high-dose pyridoxine supplementation can control seizures, but about 75% of patients still suffer from intellectual disabilities and/or developmental delay symptoms. Given that pyridoxine treatment does not fully address the issues faced by PDE patients, it is imperative to investigate the potential mechanisms by which Aldh7a1 deficiency impairs brain homeostasis after seizure control.
In a recent study, a PDE mouse model was first constructed by GUO Weixiang's group using Aldh7a1f/f::Nestin-Cre mice, which exhibited clinical symptoms of the disease.
Hippocampal neurogenesis is a unique process in the mammalian brain, where neural stem cells (NSCs) continuously generate new neurons through proliferation, differentiation, and migration, integrating them into existing circuits. This process is crucial for maintaining brain homeostasis and plasticity and is closely associated with cognitive functions such as learning, memory, and emotional regulation.
Recent studies have suggested that amino acids and their metabolic products play critical roles in cellular metabolism during adult neurogenesis, and disruptions in amino acid metabolism severely impair adult neurogenesis and cognitive abilities. Therefore, exploring the regulatory mechanisms of lysine and its metabolic products on hippocampal neurogenesis may provide insights into how PDE disrupts brain homeostasis.
To further investigate the role of ALDH7A1 in adult neurogenesis, the researchers generated Aldh7a1f/f::Nestin-CreERT2::Ai14 (Aldh7a1-iKO) mice. Although Aldh7a1-iKO mice did not exhibit spontaneous seizures, they still showed significant impairments in hippocampus-dependent cognitive tests. Furthermore, this genotype of mice exhibited pronounced abnormalities in hippocampal neurogenesis, which could not be rescued by high-dose pyridoxine treatment.
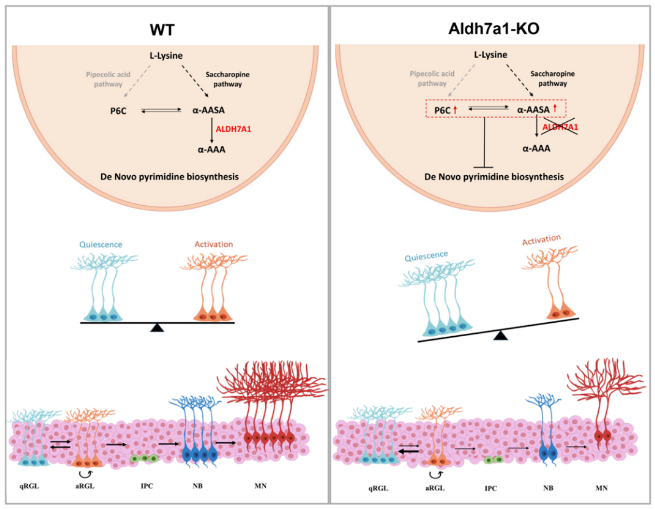
In vitro experiments revealed that only under AASA/P6C treatment did NSCs show defects in proliferation and differentiation consistent with those in ALDH7A1-deficient NSCs. Transcriptomic analysis further revealed that pyrimidine metabolism was impaired in ALDH7A1-deficient NSCs, and this impairment was independent of pyridoxine levels. Moreover, AASA/P6C administration resulted in reduced expression of genes involved in de novo pyrimidine synthesis, while exogenous pyrimidine administration could rescue defects in adult neurogenesis and cognitive impairment in Aldh7a1-deficient mice.
Therefore, ALDH7A1 deficiency leads to the accumulation of toxic metabolites in lysine metabolism, impairing de novo pyrimidine synthesis, thereby inhibiting defects in hippocampal neurogenesis and cognitive impairment, while pyrimidine administration can rescue defects in adult neurogenesis and cognitive impairment.
In summary, this study not only elucidates the potential mechanisms underlying intellectual disabilities in epilepsy patients but also provides potential strategies for treating intellectual disabilities in epilepsy patients.
The study was published in the Science Advances journal in April 2024 (DOI:10.1126/sciadv.adl2764). Dr. YAN Jianfei and Dr. WU Junjie are co-first authors of the paper, with Dr. GUO Weixiang as the corresponding author.
Working model of de novo pyrimidine synthesis’ effects on hippocampal neurogenesis and cognitive impairment (Image by IGDB)
Contact:
Dr. GUO Weixiang
Institute of Genetics and Developmental Biology, Chinese Academy of Sciences
 CAS
CAS
 中文
中文



.png)
 Working model of de novo pyrimidine synthesis’ effects on hippocampal neurogenesis and cognitive impairment (Image by IGDB)Contact:Dr. GUO WeixiangInstitute of Genetics and Developmental Biology, Chinese Academy of SciencesEmail: wxguo@genetics.ac.cn
Working model of de novo pyrimidine synthesis’ effects on hippocampal neurogenesis and cognitive impairment (Image by IGDB)Contact:Dr. GUO WeixiangInstitute of Genetics and Developmental Biology, Chinese Academy of SciencesEmail: wxguo@genetics.ac.cn