This Thursday, a group of researchers led by scientists at Institute of Genetics and Development Biology (IGDB), Chinese Academy of Sciences published a de novo assembly of an indica rice genome (Shuhui498 or R498), demonstrating how to assemble a highly contiguous and chromosome-scale near complete reference genome through an integrative strategy, which leveraged single-molecule, real time (SMRT) sequencing and pooled fosmid clone sequencing, and genetic maps and BioNano genome maps.
The R498 assembly is more continuous than the current reference genomes of
japonica rice Nipponbare (Nip, MSU7) and
Arabidopsis thaliana (TAIR10). Their results showed the genome size of
indica rice being <395 Mb. The researchers led by Dr. LIANG Chengzhi at IGDB and Dr. LI Shigui at Sichuan Agricultural University published their results in
Nature Communications (
DOI: 10.1038/ncomms15324) on May 4
th with title “Sequencing and de novo assembly of a near complete indica rice genome”.
Improving and validating genome assemblies through the application of up-to-date computational and experimental methods is an important foundation-building task for genomics-driven studies. The advent of next generation sequencing (NGS) technologies has enabled the de novo assembly of many plant genomes in the past decade. However, the genomes assembled from NGS data are usually highly fragmented draft genomes with many thousands of sequence contigs, which contain fragmented genes and collapsed or redundant repeats or chimeric contigs that confound gene functional assignment and variant detection. Even with SMRT long reads of up to 40-50 kb, the whole genome assemblies of plant genomes can still be very fragmented due to excess of repetitive sequences. Therefore, other technologies must be resorted to increase the sequence contiguity and fix assembly errors.
Compared with the commonly used scaffolding methods, the integrative method used in this study fixed many of the assembly errors during the chromosome-scale super-contig construction process. BioNano genome maps were useful for validating the high accuracy of the method and helping further correct many of the assembly errors.
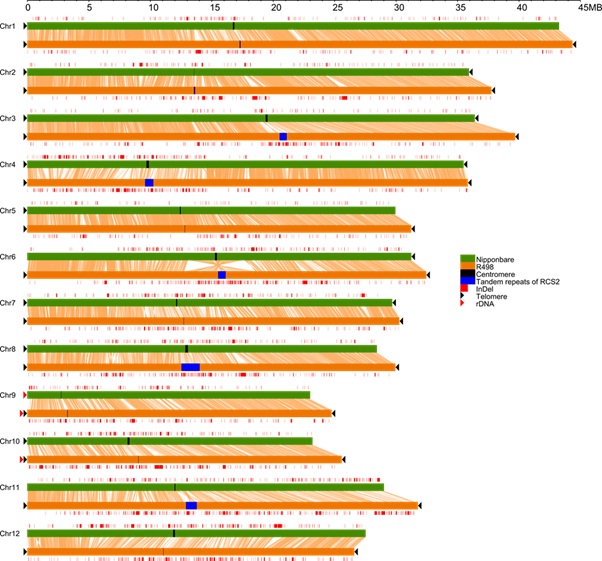
The R498 assembly showed superior quality in sequence contiguity and genome completeness. Compared with Nip, more centromere and subtelomere regions with more genes were assembled in R498, including one more nucleolus organizer region on chromosomes 10 and more LTR elements. They also assembled a complete R498 mitochondrial genome which was longer and more accurate than that of Nip. The R498 genome will serve as an ideal reference for linkage-based mutant gene identifications and genome-wide association studies in indica rice subpopulations.
The integrative method reported in this study can also be viewed as proof-of-concept on how to generate high-quality reference genomes, as it is extendable with other easily accessible technologies, e.g., by replacing genetic map with Hi-C map or replacing pooled fosmid clones with pools of DNA fragments that are generated by recently developed Chromium Genome system from 10x Genomics.
This work was supported by grants from the Chinese Academy of Sciences “Strategic Priority Research Program”, and National Natural Science Foundation of China.
 |
|
Figure 1: Whole genome comparison of R498 and Nipponbare showing end-to-end completion of each R498 chromosome and structural variations between them. (Image by IGDB) |
Contact:
Dr. LIANG Chengzhi

