Recently, Chinese scientists reported a high-quality de novo soybean genome for a Chinese soybean accession “Zhonghuang 13”. This genome and a subsequently established comprehensive gene co-expression network facilitate important agronomic genes mining and provide valuable information for future soybean elite cultivar improvement.
Soybean (Glycine max [L.] Merr.) is one of the most important crops, providing more than half of global oilseed production and more than a quarter of the world’s protein for food and animal feed. Studies have indicated that the cultivated soybean was domesticated in China approximately 5,000 years ago and then disseminated worldwide. During the introduction and dissemination process, soybean has undergone strictly genetic bottlenecks, resulting in the accessions from different geographic areas possibly exhibiting high genetic diversity.
The current soybean reference genome was sequenced from Williams 82, which is a cultivar domesticated in America. Asia is one of the largest soybean planting and consuming areas, its soybean production is essential for global food security. A high-quality reference genome is crucial for functional analysis of a species. Therefore, it is necessary to assemble a new high-quality soybean genome from Asian soybean accessions to facilitate Asia soybean functional genomics study and elite cultivar improvement.
Biologists from the Institute of Genetics and Developmental Biology, Chinese Academy of Science, University of Science and Technology of China, Jiangsu Academy of Agricultural Sciences, and Berry Genomics Corporation assembled a new soybean genome (Gmax_ZH13) for a Chinese soybean accession “Zhonghuang 13” by a combination of SMRT, Hi-C and optical mapping data.
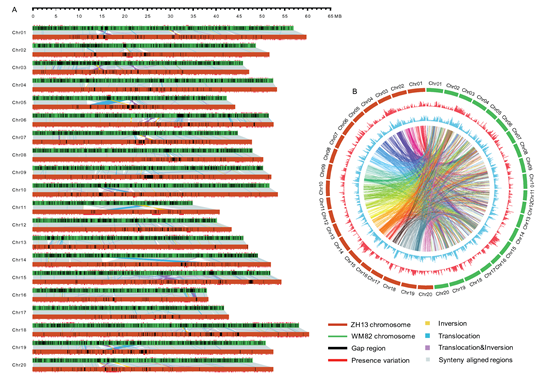
The assembled Gmax_ZH13 genome is 1.025 Gb in length with a contig N50 of 3.46 Mb and a scaffold N50 of 51.87 Mb. Comparing to the previously reported soybean reference genomes, the quality of this new genome is significantly improved which has longer total sequence length, higher contig N50 and scaffold N50, and fewer gaps. So far, it is one of the best contiguous plant genomes that have been reported. Through comparison genome analyses, the researchers identified large number of genetic variations between this genome with a commonly used soybean reference genome (Glycine_max_v2.0), including 1,404 translocations, 161 inversions, 1,233 translocation & inversions, 505,506 indels(1-99 bp) and 17,409 accession specific insertions (>=100 bp).
Through comprehensive analyses, a total of 36,429 transposable elements and 52,051 protein coding genes were annotated in the new genome. QTL and GWAS are useful approaches to investigate the loci controlling traits. However, it is difficult to identify the causal gene directly from QTL and GWAS because they usually result in large candidate regions.
Gene co-expression network is a powerful approach to explore gene regulatory relationships and to predict gene function. A gene co-expression network may assist important agronomic genes mining when combing with QTL and GWAS. To test this hypothesis, the researchers established a GGM gene co-expression network based on the annotated genes of Gmax_ZH13 using transcriptome datasets from 1,978 soybean RNA-seq that deposited in the NCBI Sequence Read Archive. The gene co-expression network helps the researchers to mine a number of candidate genes that controlling agronomically traits, such as for flowering time and linoleic acid content. Furthermore, the functions of SoyZH13_16G177400, a candidate for soybean flowering time, and SoyZH13_02G207800, a candidate gene for soybean linoleic acid content were confirmed by correlating their haplotype and phenotype information in a previously re-sequenced nature population.
This high quality Chinese soybean genome and its sequence analysis will provide valuable information for soybean improvement in the future.
This work has been published online in Science China Life Science entitled “De novo assembly of a Chinese soybean genome”.
The research was supported by the National Natural Science Foundation of China (91531304, 31525018, 31370266, 31788103), the “Strategic Priority Research Program” of the Chinese Academy of Sciences (XDA08000000), and the State Key Laboratory of Plant Cell and Chromosome Engineering (PCCE-KF-2017-03).
Figure. Genomic comparison between Gmax_ZH13 and Glycine_max_v2.0 (Image by IGDB)
Contact
QI Lei
Institute of Genetics and Developmental Biology, Chinese Academy of Science