Identification of differentially expressed genes (DEGs) is a fundamental step for many biomedical studies. RNA-sequencing is the most popular approach for this purpose and is widely used. For example, two algorithms for RNA-sequencing differential expression (DE) analysis, edgeR and DESeq2, have been cited over 10,000 times.
DE Analysis with inadequate replicates affects the accuracy and has the potential to invalidate many conclusions arrived at in such a manner. Biostatisticians have long encouraged the use of more biological replicates to achieve sufficient statistical power to call DEG correctly. However, due to limitations of current RNA-sequencing profiling methods, including relatively high cost and complex processing, most studies used only 2-3 replicates. At this level of replication, only the most strongly changing genes can be identified. Therefore, the lack of practical experimental approaches prevents biologists from employing adequate replicates to achieve sufficient power in DEG discovery studies.
Recently, a team lead by Dr. TU Qiang at the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, developed a practical approach Decode-seq (Differential Expression analysis by barCODEd SEQuencing) to overcome the current limitations in the RNA-seq DE analysis and enable more accurate DEG discovery.
Decode-seq uses molecular barcodes to profile a few dozens of samples in one library simultaneously, which significantly reduces the time and cost of library preparation. It enriches 5′ cDNA ends for sequencing, which reduces sequencing depth required compared to the full-length sequencing and overcomes the difficulty of 3′ end sequencing. Decode-seq libraries are compatible with standard Illumina sequencing settings. Therefore, it does not require customized sequencing cycle numbers or primers, and users do not have to devote the entire flow cell or lane for sequencing.
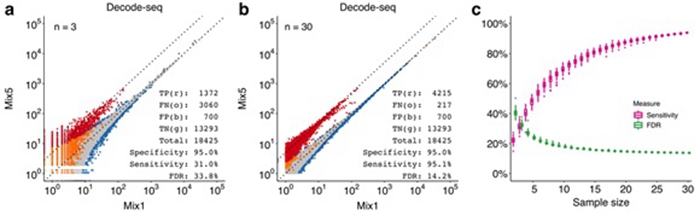
These features greatly reduce the profiling cost to about 10% of the traditional RNA-seq method. In the validation experiments, when the replicate number increased from 3 to 30, the sensitivity increased from 31% to 95%, and the false discovery rate dropped from 34% to 14%. Decode-seq is also compatible with diminutive amounts of RNA.
Researchers applied this method in analyzing medaka fish at the early stage of sex determination with 30 pairs of replicates. They discovered multiple novel sexually dimorphically expressed genes, some of which are required for germ cell development.
Briefly, increasing replicates significantly improves the performance of DE analysis, and Decode-seq provides a practical approach for this purpose. Unless samples are precious, full-length sequencing is necessary, or underpower is not an issue, there are few reasons to use inadequate replicates. Therefore, the scientists appeal that the common practice of using three replicates in DE analysis should be avoided if possible. The experimental protocol and computational pipeline are available from
the homepage of TU’s lab.
The paper entitled “
Decode-seq: a practical approach to improve differential gene expression analysis” was published online in
Genome Biology on March 23, 2020 (
DOI: 10.1186/s13059-020-01966-9). This work was supported by the National Natural Science Foundation of China and the Chinese Academy of Sciences.
Figure: Performance evaluation of Decode-seq. (a) Differential expression analysis with 3 pairs of replicates. True positive/TP, red dots; false negative/FN, orange dots; true negative/TN, gray dots; false positive/FP, blue dots. (b) DE analysis with 30 pairs of replicates. The sensitivity increased to 95.1%, and the false discovery rate dropped to 14.2%. (c) DE performance related to replicate number calculated by random downsampling of 30-pair data. Sensitivity and false discovery rate were improved dramatically when the number of replicates increased. (Image by IGDB)
Contact:
QI Lei
Institute of Genetics and Developmental Biology, Chinese Academy of Sciences