Prime editing (PE), a “search-and-replace” CRISPR-based genome editing technique, has great potential for gene therapy and agriculture. It can introduce desired base conversions, deletions, insertions, and combination edits into target genomic sites. Prime editors have been successfully applied in animals and plants, but their off-target effects, which can be a major hindrance to real-life application, have not been thoroughly evaluated until now.
Prof. GAO Caixia from the Institute of Genetics and Developmental Biology (IGDB) of the Chinese Academy of Sciences (CAS) and her research team recently performed a comprehensive and genome-wide analysis of the off-target effects of PEs in rice plants.
Off-target effects are in principle of two types: guide RNA (gRNA)-dependent and gRNA-independent. The first result from similarities between target and off-target sequences and the second from the activity of CRISPR-based tools, such as deaminase, at non-target positions in the genome.
The researchers first measured editing frequencies using pegRNAs with primer binding sites (PBSs) or spacers containing mismatches of the chosen target sequence, and found that mismatches located in seed sequence regions of the spacer (near the PAM) and near the nicking site of nCas9 (H840A) at the PBS greatly reduced the frequency of PE implying high editing specificity. They also evaluated the frequencies of editing by 12 pegRNAs at 179 endogenous off-target sites containing mismatches, and confirmed that editing rates were extremely low (0.00%~0.23%). Thus, designing pegRNAs with homology to fewer off-target sites is necessary.
The gRNA-independent effects induced by ectopic expression of functional elements in the CRISPR-based tools (which have been detected with some base editors) are not predictable by in silico methods. Gao et al. therefore used whole-genome sequencing to investigate whether ectopic expression of the prime editors induced undesired edits at the genome-wide level. They delivered PE constructs with or without pegRNA expression cassettes into rice calli via Agrobacterium-mediated transformation and obtained regenerated T0 plants (the PE group). They found the number of single nucleotide variants and indels (small insertions/deletions) in the PE group was not significantly higher than in the control group (expressing Cas9 nickase). Moreover, mutation type analysis and mutation distribution analysis further demonstrated that the PE and control groups did not differ significantly. This result indicated that the PE system did not induce significant numbers of genome-wide pegRNA-independent off-target edits in plants.
Since M-MLV RT is a core element of the PE system, it seemed possible that overexpressing M-MLV RT might interfere with natural reverse transcription mechanisms in the cell. The researchers therefore evaluated the activities of retrotransposons and telomerase by analyzing the number of copies of the OsTos17 retrotransposon and the fidelity of telomeres, and found no effect of M-MLV reverse transcriptase on either parameter. They also evaluated the possibility that over-expression of RT might increase the risk of random reverse transcription of mRNAs and insertion of the products into the rice genome. Hence, they looked for pegRNA and mRNA insertions but detected no such events, further indicating that the M-MLV reverse transcriptase in PEs does not have nonspecific effects in plant cells. In summary, a systematic assessment demonstrated that prime editors are highly specific in plants.
This work, entitled "Genome-wide specificity of prime editors in plants," was published in
Nature Biotechnology (
DOI: 10.1038/s41587-021-00891-x)
online on April 15. This research was supported by the National Natural Science Foundation of China, the National Key Research and Development Program of China, and the Strategic Priority Research Program of CAS.
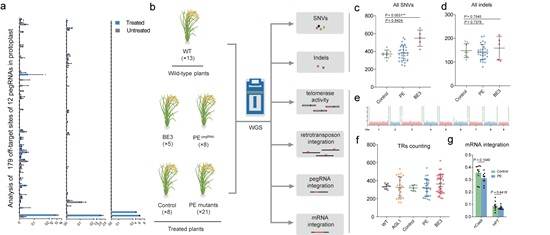
Figure. Analysis of the genome-wide specificity of prime editors in plants. (Image by IGDB) a, PE rates at endogenous on-target and off-target sites. b, Experimental design and work flow of whole-genome sequencing. c, d, Numbers of SNVs (c) and Indels (d) identified by WGS in the control, PE and base editor (BE3) groups. e, Genome-wide landscape of the distribution of telomere repeats in the Zhonghua11 genome. f, Comparison of number of reads with more than five TRs per million raw reads in the WT, AGL1, control, PE and BE3 groups. g, Comparison of the percentages of reads in the Cas9 and HPT coding regions versus in the whole T-DNA fragment.
Contact:
Dr. GAO Caixia
Institute of Genetics and Developmental Biology, Chinese Academy of Sciences
Email: cxgao@genetics.ac.cn

